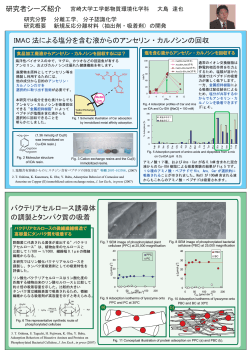
Molecular Simulation, 2014 Vol. 40, Nos. 7 – 9, 516–536, http://dx.doi.org/10.1080/08927022.2013.832247 Molecular simulations in metal –organic frameworks for diverse potential applications Jianwen Jiang* Department of Chemical and Biomolecular Engineering, National University of Singapore, 117576, Singapore Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 (Received 25 June 2013; final version received 28 July 2013) As a new class of intriguing nanoporous materials, metal –organic frameworks (MOFs) have been considered for diverse potential applications. The number of MOFs synthesised to date is extremely large; thus, experimental testing alone is economically expensive and practically formidable. With rapidly growing computational resources, molecular simulation has become an indispensable tool to characterise, screen and design MOFs. Our research group has conducted comprehensive simulation studies in MOFs for carbon capture, hydrocarbon separation, alcohol adsorption and biofuel purification, water treatment, bio- and chiral separation and drug loading; furthermore, mechanical moduli, structural transition and thermal conductivity have also been examined. These systematic simulation studies are summarised in this review to demonstrate that simulation at a molecular level can secure the quantitative interpretation of experimental observation, provide the microscopic insight from bottom-up and facilitate the development of new MOFs. Keywords: metal – organic frameworks; potential applications; molecular simulations Jianwen Jiang’s research expertise is in computational materials modeling and statistical thermodynamics. His current research activities are focused on porous and membrane materials for energy, environmental, and pharmaceutical applications such as carbon capture, water treatment, biofuel purification, and drug delivery. He has published over 140 technical manuscripts in peerreviewed journals and a number of invited reviews and book chapters. He is on the editorial boards of Advances in Materials Research and ISRN Nanotechnology. 1. Introduction In the last decade, metal–organic frameworks (MOFs) have emerged as a new family of nanoporous materials.[1] Assembled by direct bonding between metal clusters and organic linkers, the structures of MOFs are largely predictable. In contrast to conventional zeolitic materials consisting of tetrahedral building blocks, MOFs can be synthesised from various inorganic clusters (e.g. squareshaped, trigonal, tetrahedral and octahedral) and organic linkers (e.g. carboxylates, imidazolates and tetrazolates). Consequently, MOFs possess a wide range of surface area, pore size and volume. With these salient features, MOFs have been considered versatile materials for diverse potential applications,[2–4] as illustrated in Figure 1, ranging from gas storage, separation, water desalination, biofuel purification to catalysis, drug delivery, etc. Indeed, MOFs have been identified as a topical area in materials science and technology because of their foreseeable implications.[5] Thousands of MOFs have been synthesised, and several of them are commercially available under the trade name BasoliteTM (e.g. Cu-BTC, Fe-BTC, MIL-53 and *Email: [email protected] q 2014 Taylor & Francis ZIF-8). Enormous experimental studies have been reported on the characterisation and applications of various MOFs, primarily on H2 storage [6] and CO2 capture.[7] However, the number of MOF materials synthesised is extremely large and in principle unlimited; thus, experimental testing and screening of ideal MOFs from infinite possible candidates are time-consuming and practically infeasible. With rapidly growing computational resources, molecular simulation has become an indispensable tool and plays an increasingly important role in materials science and engineering. Sophisticated simulation at an atomistic/molecular level provides microscopic insight that is experimentally intractable, and elucidates underlying physics from bottom-up. In addition, simulation can be used to secure the fundamental interpretation of experimental observation and to establish the structure – function relationship to guide the rational selection and design of novel materials.[8] To date, most simulation studies in MOFs have been focused on gas storage and separation, particularly the Molecular Simulation CO2 Capture Gas Storage Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 Removal of Toxics Water Desalination Figure 1. 517 Biofuel Purification Drug Delivery Catalyts or Supports Synthetic Channels (Colour online) Diverse potential applications of MOFs. storage of low-carbon footprint energy carriers (e.g. H2) and the separation of CO2-containing gas mixtures for carbon capture.[9 –14] In our research group, comprehensive simulation studies have been conducted in MOFs for a wide range of diverse potential applications such as carbon capture, hydrocarbon separation, alcohol adsorption and biofuel purification, water treatment, bio- and chiral separation, drug loading, as well as mechanical moduli, structural transition and thermal conductivity. As invited by the Editor, these systematic simulation studies are summarised in this review. 2. Simulation methodology The atomic coordinates of MOFs in simulation are usually adopted from experimentally crystallographic data. It should be noted that some crystallographic data contain the coordinates of solvent or ligand molecules or diffraction defects, and cannot be directly input into simulation. For this case, the crystallographic data are required to be modified so as to properly represent the crystalline structures. Unless an interfacial region is examined, simulation box is mimicked by periodic boundary conditions and surrounded by its replicated images. Thus, simulation system can be considered to be an infinitely large crystal. In most simulation studies, the frameworks are assumed being fixed. Such a treatment is acceptable for relatively rigid MOFs, and indeed, it has two advantages: first, there is no need to consider the intra-framework interactions and second, the guest –framework interactions can be pre-tabulated prior to simulation. Therefore, it is crucial to accurately describe the guest – framework interactions that exclusively determine the reliability of simulation. The guest– framework interactions can be represented by long-range electrostatic potential and shortrange van der Waals potential. For the electrostatic potential, atomic charges in MOFs need to be evaluated, and several methods have been proposed in the literature, such as quantum mechanics (QM)-based electrostatic potential method,[15,16] connectivity-based atom contribution method [17] and extended charge equilibration method.[18] For the van der Waals potential, QM methods can be used to derive the potential parameters in two steps. Firstly, the interaction energies between guest and framework are calculated by QM at various representative positions and orientations. Secondly, an analytic potential function is adopted to fit the interaction energies.[19,20] To accurately calculate the interaction energies, an appropriate level of QM method is needed. The most costeffective method is density functional theory (DFT). However, DFT has deficiency in describing the non-local dispersion forces induced by electron correlations. In principle, wave function-based coupled-cluster and Møller –Plesset perturbation methods are more accurate, but they are computationally very demanding particularly for large systems. As an alternative, molecular mechanical (MM) force fields can be used to describe the van der Waals potential between guest and framework. For example, universal force field (UFF) [21] and DREIDING [22] have been widely used for MOFs. The potential parameters in these force fields are usually derived from experimental data over a limited range of conditions. Although useful Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 518 J. Jiang information can be obtained from them, the empirical nature may lead to inaccurate or incorrect predictions. Consequently, they should be used with caution. A number of MOFs exhibit flexible/dynamic structures upon stimuli (e.g. temperature, pressure, sorption and so on). In contrast to rigid MOFs, the intra-framework interactions should be incorporated to mimic framework flexibility, including bond stretching, bending and torsional potentials. These potentials can also be determined from QM methods, but computationally expensive; a more common way is to use MM force fields. With a proper description of molecular interactions, simulation can be performed using Monte Carlo (MC), molecular dynamics (MD) or a hybrid MC and MD. [23,24] In MC simulation, representative configurations are randomly generated. Thus, MC simulation can be performed with physically unnatural trial moves, e.g. a jump from one position to the other or a random insertion/ deletion. MC simulation is well suited to investigate adsorption in MOFs. Particularly, grand canonical MC (GCMC) simulation is used very widely to simulate adsorption isotherms and other thermodynamic properties (e.g. Henry’s constant, isosteric heat and so on). Based on the Newton’s second law of motion, MD simulation mimics the natural pathway of molecular motion to sample successive configurations. At each time during MD simulation, the forces between molecules are calculated, then the equations of motion are solved numerically and finally, velocities and positions are updated. The time step chosen should be sufficiently small; thus, the total energy of system is conserved. In addition to thermodynamic properties that can be simulated by MC method, transport properties such as diffusivity and conductivity are usually determined from MD method. For either method, simulation is anticipated to run for a sufficient long time to assure that equilibration or steady state is reached; thereafter, ensemble averages can be estimated. 3. Diverse potential applications As listed in Table 1, our group has systematically conducted a series of simulation studies in MOFs for diverse potential applications including carbon capture (in both MOF adsorbents and membranes), hydrocarbon separation, alcohol adsorption and biofuel purification, water treatment, bio- and chiral separation and drug loading. Moreover, we have also developed force fields incorporating framework flexibility to describe mechanical moduli, structural transition and thermal conductivity. 3.1 Carbon capture A vast amount of CO2 emissions have been produced by the combustion of fossil fuels, as estimated to be 30 gigatons per year worldwide.[63] Carbon capture and sequestration (CCS) is critical for environmental protection and sustainable development. As a key step in CCS, CO2 is required to be captured in pre-combustion or postcombustion process. A handful of technologies have been proposed for carbon capture such as cryogenic distillation, amine scrubbing, membrane separation and sorbent adsorption.[64] Due to the occurrence of phase transition, cryogenic distillation is economically expensive. Although amine scrubbing is commercially applied in industry, regeneration step is energetically intensive. Comparatively, sorbents and membranes-based carbon capture are more efficient with lower capital cost and larger separation capability. The majority of experimental and simulation studies in MOFs have been focused on carbon capture, as summarised in several reviews.[12,65 – 67] 3.1.1 Adsorbents Synthesised MOFs are crystallites and usually tested as adsorbents for carbon capture. It is noteworthy that most of experiments examine the adsorption of pure gases (e.g. CO2, N2, CH4 and H2) due to the difficulty in measuring mixture adsorption. On the contrary, simulation can be readily used for mixtures. Therefore, quantitative understanding of mixture adsorption in MOFs has been largely based on simulation. Isoreticular MOFs. One of the earliest simulation studies we carried out was carbon capture in IRMOF-1.[25] As a prototype of MOF, IRMOF-1 consists of Zn4O as a metal cluster and 1,4-benzenedicarboxylate (BDC) as an organic linker.[68] The straight pore in IRMOF-1 possesses ˚ . Specifically, we alternating diameters of 15 and 12 A predicted the adsorption and separation of CO2 and CH4 in IRMOF-1, silicalite and C168 schwarzite at 300 K.[25] The separation of CO2/CH4 mixture is important in the sweetening of nature gas. As illustrated in Figure 2, the three materials have well-defined three-dimensional (3D) structures, of which each represents a typical class of nanoporous materials. The adsorption isotherms and isosteric heats of pure gases from simulation agree well with available experimental data. CO2 is preferentially adsorbed over CH4 in all the three adsorbents, except C168 schwarzite at high pressures. All isotherms are of type I (Langmuirian), which is the characteristic of microporous adsorbents with a pore of molecular dimension (below ˚ ). The adsorption capacities of CH4 and CO2 in 20 A IRMOF-1 are substantially larger than in silicalite and C168 schwarzite. This implies that IRMOF-1 might be a potential medium for gas storage. The predictions of mixture adsorption from the ideal adsorbed solution theory (IAST) [69] are in accord with simulation, particularly at low pressures because the IAST becomes exact in Henry’s Molecular Simulation Table 1. Summary of simulation studies in MOFs for diverse potential applications. Application CO2 capture in adsorbents Isoreticular MOFs Catenated MOFs Functionalised MOFs Ionic MOFs Moisture effects Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 CO2 capture in membranes MOF membranes Polymer/MOF membranes IL/MOF membranes Hydrocarbon separation Alkanes (C1 – C5) Xylene isomers Alcohol adsorption and biofuel purification Alcohol adsorption (C1 – C4) Biofuel purification Dilute biofuel purification Water treatment Water desalination Removal of toxic ions (Pd2þ) Removal of organics (DMSO) Bio- and chiral separation and drug loading Bio-separation of amino acids Chiral separation Ibuprofen loading Mechanical moduli, structural transition and thermal conductivity Mechanical moduli Structural transition Thermal conductivity MOF examined Reference IRMOF-1, -13, -14 IRMOF-13, PCN-6 bio-MOF-11, ZTF, F-MOFs, MIL-101 soc-MOF, rho-ZMOF, rht-MOF, Li-MOF soc-MOF, rho-ZMOF, rht-MOF, bio-MOF-11, MIL-101, Zn(bdc)(ted)0.5 [25,26] [26] [27 – 32] [33 – 37] [27,33,35,38 –40] IRMOF-1, ZIF-95, Zn(bdc)(ted)0.5 PBI/ZIF-7 [BMIM][X]/IRMOF-1 (X ¼ [Tf2N], [BF4], SCN]), [BMIM][SCN]/ZIF-71, [BMIM][SCN]/ rho-ZMOF [41 – 43] [44] [45 – 47] IRMOFs, PCNs MIL-101 [48,49] [50] Zn(bdc)(ted)0.5, rho-ZMOF, Zn4O(bdc)(bpz)2, ZIF-71, ZIF-8 rho-ZMOF, Zn4O(bdc)(bpz)2 hydrophobic MOFs and ZIFs [40,51– 53] [52] [54] ZIF-8 membrane rho-ZMOF ZIF-71, Zn4O(bdc)(bpz)2, Zn(bdc)(ted)0.5 [55] [56] [57] MIL-101 Zn2(bdc)(L-lac)(dmf) MIL-101, UCMC-1 [50] [58] [59] ZIF-8, ZIF-7, PBI/ZIF-7 ZIF-8 ZIF-8, MOF-5 law regime. At high pressures, however, the IAST either underestimates or overestimates the simulated results. This is attributed to the assumption used in the IAST, i.e. the adsorbed phase of mixture is assumed to be ideally mixed. Though IRMOF-1 has a significantly higher adsorption capacity than silicalite and C168 schwarzite, CO2/CH4 selectivity is close in the three adsorbents. Substituting the BDC linker in IRMOF-1 by a longer linker namely pyrene dicarboxylate, IRMOF-14 can be Figure 2. 519 [60,61] [60,61] [62] ˚ .[68] generated with pore diameters of 20.1 and 14.7 A From simulation, we observed that CO2/CH4 selectivity in IRMOF-14 is lower than in IRMOF-1.[26] This is because the larger pore in IRMOF-14 is less favourable for separation. The presence of electrostatic interactions between CO2 and framework was found to cause an increase in CO2 adsorption and a decrease in CH4 adsorption; consequently, CO 2/CH 4 selectivity is enhanced. (Colour online) IRMOF-1, silicalite and C168 schwarzite.[25] J. Jiang Catenated MOFs. To improve carbon capture in MOFs, several strategies have been proposed such as using catenated, functionalised and ionic frameworks.[13] We performed simulation to investigate the separation of CO2/ CH4 mixture in IRMOF-13 and IRMOF-14 at 300 K.[26] Despite identical metal oxide and organic linker, IRMOF13 has a catenated framework and differs from noncatenated IRMOF-14. As a result, pore diameters in ˚ , smaller than in IRMOF-14. IRMOF-13 are 12.4 and 8.7 A CO2 adsorption at low pressures in IRMOF-13 is greater than in IRMOF-14. The reason is that the formation of constricted pores by catenation leads to a larger degree of potential overlap and a stronger affinity for adsorbate. Due to the smaller pore volume in catenated framework, however, the opposite is true at high pressures. As shown in Figure 3, IRMOF-13 has a higher selectivity than noncatenated counterpart for CO2/CH4 mixture. The effect of electrostatic interaction on the selectivity is larger in IRMOF-13 than in IRMOF-14. PCN-60 and PCN-6 were also examined for the separation of CO2/CH4 mixture.[26] Consisting of paddlewheel coppers linked with 4,40 ,400 -s-triazine-2,4,6-triyltribenzoate, PCN-60 is non-catenated and iso-structural to Cu-BTC.[70] The square pore in PCN-60 possesses a large ˚ £ 21.44 A ˚ . In contrast, PCN-6 is catenated size of 21.44 A 0 ˚. counterpart of PCN-6 with a triangular pore of 9.2 A 0 PCN-6 and PCN-6 exhibit adsorption behaviour similar to IRMOF-14 and IRMOF-13, respectively. CO 2/CH4 selectivity in catenated PCN-6 is higher than in noncatenated PCN-60 . Functionalised MOFs. Functionalisation is a commonly used method to tailor material properties and has been applied to tune MOFs. We simulated the separation of 7 2/CH4 IRMOF-13 5 SCO Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 520 3 IRMOF-14 1 0 1000 2000 3000 4000 5000 P (kPa) Figure 3. (Colour online) CO2/CH4 selectivity in IRMO-13 and IRMOF-14.[26] CO2/H2 and CO2/N2 mixtures in bio-MOF-11 with adenine as a bio-linker.[27] The simulated and experimental adsorption isotherms of pure CO2, H2 and N2 are in a perfect agreement. As attributed to the presence of multiple Lewis basic sites (amino and pyrimidine groups) and nano-sized channels, bio-MOF-11 has a larger adsorption capacity and a higher selectivity than many zeolites, activated carbons and MOFs. The study provides microscopic insight into the adsorption mechanism in bioMOF-11 and suggests that bio-MOF-11 might be interesting for pre- and post-combustion carbon capture. By combining experiment and simulation, we examined CO2 adsorption in amino-functionalised zeolitic tetrazolate framework (ZTF).[28] The Zn · · · Zn distance ˚ , extended by replacing oxide ions with in ZTF is 5.9 A ˚, tetrazolate linkers. ZTF possesses a pore diameter of 4.5 A with a diamond (dia) topology and extended 3D structure. CO2 capacity was experimentally measured to be 5.6 mmol/g at 273 K, representing one of the highest values. The high CO2 capacity was attributed to the narrow pores, exposed ZNH2 functionality and free tetrazole nitrogen. Good agreement was found between the experimental isotherm and simulation. Furthermore, we investigated CO2 adsorption in two iso-structuralinterpenetrated amino-functionalised Cd-ANIC-1 and Co-ANIC-1 (ANIC: 2-amino-4-pyridine carboxylic acid).[29] The two MOFs adopt dia topology with amino groups lined along the pores. High CO2 uptake was observed experimentally and validated by simulation, as attributed to the presence of Lewis basic amino groups and framework interpenetration. We synergised ab initio calculation, molecular simulation and breakthrough prediction to examine postcombustion carbon capture at 303 K in MIL-101 functionalised by a series of groups (X ¼ ZNH2, ZCH3, ZCl, ZNO2 and ZCN).[32] MIL-101 is a chromium terephthalate-based mesoscopic MOF and one of the most porous materials reported to date.[71] It is stable in air and boiling water, and its structure is not altered in organic solvents or solvothermal conditions. In low-pressure regime, CO2 uptake and isosteric heat increase in the order of MIL-101 , MIL-101-CN , MIL-101-NO 2 , MIL-101-Cl , MIL-101-CH3 , MIL-101-NH2. Such an order follows the strength of binding energies between CO2 and functional groups calculated by ab initio method. However, the effect of functional group is marginal for N2 adsorption. As shown in Figure 4, CO2/N2 selectivity is enhanced by functionalisation, following MIL101 , MIL-101-CN , MIL-101-CH3 , MIL-101-NO2 , MIL-101-Cl , MIL-101-NH2. At infinite dilution, the enhancement of selectivity is 2.5 times, from 16 in parent MIL-101 to 40 in MIL-101-NH2. The predicted breakthrough time is extended by functionalisation, and the longest breakthrough time in MIL-101-NH2 is 2 times that in parent MIL-10. The multi-scale modelling study Molecular Simulation 60 40 Mole fraction of outlet CO2 50 S(CO2/N2) 0.15 MIL-101-NH2 MIL-101-Cl MIL-101-CH3 MIL-101-NO2 MIL-101-CN MIL-101 30 20 0 20 40 60 80 MIL-101 MIL-101-NO2 MIL-101-CN MIL-101-Cl MIL-101-CH3 MIL-101-NH2 0.12 0.09 0.06 0.03 0.00 100 0 20 40 Ptotal (kPa) 60 80 100 120 140 160 Dimensionless time Figure 4. (Colour online) (a) CO2/N2 selectivity in MIL-101-X and (b) mole fraction of outlet CO2 through a fixed bed packed with MIL-101-X. The inlet gas mixture consists of 15 kPa CO2 and 85 kPa N2.[32] suggests that the performance of carbon capture in MIL101 can be considerably improved by functionalisation. eight-membered ring for the adsorption of CO2/CH4 mixture at a total pressure of 500 kPa. CO2 molecules are observed to bind preferentially with Naþ ions, as also demonstrated by the radial distribution functions in Figure 5(b). The predicted CO2/CH4 selectivity is significantly higher than in most MOFs. By artificially switching off the charges in Na-rho-ZMOF, the selectivity decreases substantially. This suggests that electrostatic interaction is crucial for the high selectivity in Na-rhoZMOF. We further investigated the separation of CO2/H2 mixture in three ionic MOFs (rho-ZMOF, soc-MOF and rht-MOF).[33 –35] Similar to CO2/CH4 mixture in rhoZMOF, the predicted CO2/H2 selectivity in the three ionic MOFs is much higher than in non-ionic MOFs. Particularly noteworthy is that the selectivity behaves differently in the three ionic MOFs with varying free volume and charge Ionic MOFs. In a porous material, the presence of chargebalancing non-framework ions can enhance the interactions with guest molecules, which in turn increase adsorption capacity and selectivity. We simulated the separation of CO2/CH4, CO2/N2 and CO2/H2 mixtures in Na-rho-ZMOF at 298 K.[34] The rho-ZMOF possesses a widely open anionic framework and charge-balancing doubly protonated 1,3,4,6,7,8-hexahydro-2H-pyrimido [1,2-a]pyrimidine (HPP).[72] The HPP ions can be exchanged with other cations, e.g. Naþ ions. The simulation revealed that CO2 is predominantly adsorbed over other small gases due to the strong electrostatic interactions with Naþ ions and ionic framework. Figure 5 (a) demonstrates the typical locations of CO2 molecules in (a) (b) 1000 5 + Na - CO2 g (r) 4 SCO2/CH4 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 10 521 3 2 + Na - CO4 1 100 0 2 4 6 8 10 r (Å) 10 0 500 1000 1500 2000 2500 3000 P (kPa) Figure 5. (Colour online) Adsorption of equimolar CO2/CH4 mixture in Na-rho-ZMOF. (a) Locations of CO2 molecules in eightmembered ring. Naþ ions and CO2 molecules are represented by balls and sticks, respectively. (b) CO2/CH4 selectivity. The inset shows the radial distribution functions.[34] 522 J. Jiang decreases due to two factors. First, the adsorption sites are heterogeneous and adsorbate molecules start to occupy less favourable sites at high pressures. Second, H2 is smaller than CO2 and can pack into the partially filled cages more easily with increasing pressure. 1e+5 Na K Rb Cs Mg Ca Al SCO2/H2 1e+4 1e+3 1e+2 1 10 100 1000 Figure 6. CO2/H2 selectivity in Naþ, Kþ, Rbþ, Csþ, Mg2þ, Ca2þ and Al3þ-exchanged rho-ZMOFs.[37] density. The fractional free volume increases in the order of rho-ZMOF , soc-MOF , rht-MOF, but the charge density increases in the opposite order. Consequently, rhoZMOF has the smallest porosity and the largest charge density, and thus, exhibits the highest selectivity, followed by soc-MOF and rht-MOF. The Naþ ions in Na-rho-ZMOF can be exchanged with other ions. To assess the effect of such exchange, the separation of CO2/H2 mixture was simulated in rho-ZMOFs exchanged with various cations including Naþ, Kþ, Rbþ, Csþ, Mg2þ, Ca2þ and Al3þ.[37] Figure 6 shows CO2/H2 selectivities in the seven rho-ZMOFs versus the total pressure. At a given pressure, the selectivity increases as Csþ , Rbþ , K þ , Na þ , Ca 2þ , Mg2þ < Al3þ. This is because the electric field of cation increases with increasing charge-to-diameter ratio, and hence, the interaction with CO2 is enhanced. With increasing pressure, the selectivity in each rho-ZMOF (a) 70 (b) 20 CO2 /H2 /CO/CH4 (15:75:5:5) 60 15 NO3–H2O g(r) SCO2/H2 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 P (kPa) Moisture effects. Moisture usually exists in gas mixtures and can adversely affect separation performance. We have systematically examined the moisture effects on the adsorption and separation of CO2 in various MOFs, and observed four intriguing effects depending on the nature of framework. (1) In ionic rht-MOF, the presence of H2O substantially reduces CO2/H2 selectivity as shown in Figure 7(a).[35] The radial distribution functions in Figure 7(b) reveal that H2O is much more strongly adsorbed onto NO2 3 ions than CO2. Consequently, the interaction between CO2 and NO2 3 ion is substantially shielded by H2O, which in turn reduces CO2 adsorption and CO2/H2 selectivity. A similar significant effect of H2O on CO2/CH4 separation was also observed in rho-ZMOF.[38] It is therefore important to remove H2O before carbon capture in these ionic MOFs. (2) Though with ionic framework, soc-MOF exhibits different behaviours from rht-MOF and rho-ZMOF.[33] The non-framework NO2 3 ions in soc-MOF are located in carcerand-like capsules connected via narrow windows with dimensions of ˚ £ 5.946 A ˚ . In the presence of a small amount 7.651 A H2O, CO2/H2 selectivity in soc-MOF increases at low pressures as a consequence of the promoted adsorption of CO2 by H2O bound onto exposed indium atoms. This is because H2O interacts preferentially with the readily accessible indium atoms rather than NO2 3 ions in the capsules, and thus, the bound H2O molecules act as additional sites for CO2 adsorption. With increasing H2O at 50 CO2/H2 /CO/CH4/H2O (15:75:5:5:0.1) 40 10 5 NO3–CO2 NO3–H2 0 30 0 1000 2000 3000 P (kPa) 4000 5000 2 3 4 5 6 7 8 9 r (Å) Figure 7. (Colour online) (a) CO2/H2 selectivity in CO2/H2/CO/CH4 mixture (15:75:5:5) and CO2/H2/CO/CH4/H2O mixture (15:75:5:5:0.1). (b) Radial distribution functions between NO2 3 and adsorbates in CO2/H2/CO/CH4/H2O mixture (15:75:5:5:0.1). The inset illustrates a simulation snapshot.[35] high pressures, however, H2O competitively replaces CO2 and the selectivity decreases. (3) In non-ionic MIL-101, terminal H2O molecules enhance the adsorption of CO2 and CH4 at low pressures particularly for CO2.[39] The reason is that the terminal H2O molecules act as additional adsorption sites and CO2 interacts more strongly with them than CH4. At high pressures, however, the terminal H2O molecules reduce the free volume of MIL-101 and lead to a smaller adsorption capacity compared with dehydrated MIL-101. CO2/CH4 selectivity is slightly higher in hydrated MIL-101.(4) In a hydrophobic MOF namely Zn (bdc)(ted)0.5 (bdc ¼ benzenedicarboxylate and ted ¼ triethylenediamine), H2O adsorption is vanishingly small as verified by both experiment and simulation, attributed to the highly hydrophobic nature of Zn(bdc)(ted)0.5.[40] The bdc and ted linkers surround metal oxides; thus, Zn(bdc) (ted)0.5 interacts with H2O very weakly. Consequently, the adsorption and selectivity of CO2/CH4 mixture remain nearly identical in the absence or presence of H2O. 3.1.2 Membranes The above-discussed simulation studies for carbon capture are based on MOF adsorbents. There is an increasing interest in the application of MOF membranes for gas separation, though it is not in a mature stage.[73] We have also investigated MOF membranes and composite membranes for carbon capture. To calculate permeation in a membrane, both equilibrium and dynamic properties are required; thus, it is more challenging and timeconsuming to simulate. MOF membranes. The diffusion and separation of CO2 and CH4 in IRMOF-1, silicalite and C168 schwarzite at 300 K were compared by simulation.[41] As loading increases, the self-diffusivities in the three structures decrease due to steric hindrance; the corrected diffusivities remain nearly constant or decrease approximately in a linear manner depending on the type of adsorbate and structure; the transport diffusivities generally increase except for CO2 in IRMOF-1. The correlation effects reduce in the order of silicalite . C168 . IRMOF-1, opposite to the increasing hierarchy of porosity (silicalite , C168 , IRMOF-1) in the three structures. The predicted self-, corrected and transport diffusivities of CO2 and CH4 from the Maxwell –Stefan formulation are consistent with simulation results. The permselectivity is marginal in IRMOF-1, slightly enhanced in silicalite and C168 schwarzite. Overall, the three structures are not suitable for CO2/CH4 separation. By combining experiment and simulation, we examined H2/CO2 separation in a highly permeable and selective ZIF-95 membrane.[42] In the synthesised ZIF-95 membrane, single gas permeances follow the order of H2 523 . N2 . CH4 . CO2 . C3H8, which mainly corresponds to their kinetic diameters with the exception of CO2. The reason is that CO2 has a strong quadrupolar interaction with nitrogen atoms present in the linkers of ZIF-95, leading to a high adsorption capacity but lowering diffusional mobility, which results in a lower CO2 permeance than other small gas molecules (e.g. N2 and CH4). The molecular sieve performance of ZIF-95 membrane was confirmed by the separation of equimolar gas mixtures at 3258C and 1 bar. For H2/N2, H2/CH4 and H2/C3H8 mixtures, the separation factors were measured to be 10.2, 11.0 and 59.7, which exceed the corresponding Knudsen coefficients (3.7, 2.8 and 4.7). The ZIF-95 membrane shows by far the highest H2/CO2 selectivity among reported MOF and zeolite membranes. As shown in Figure 8, the performance of ZIF-95 membrane surpasses Robeson’s upper bound.[74] This study suggests that hydrothermally stable and highly permselective ZIF-95 membrane is potentially interesting for H2 purification. Furthermore, a hydrophobic Zn(bdc)(ted)0.5 membrane was tested for H2/CO2 separation.[43] Attributed to the preferential adsorption affinity for CO2, the membrane displays a high H2/CO2 permselectivity. For equimolar H2/ CO2 mixture at 1808C and 1 bar, H2 permeance of 2.65 £ 1026 mol m22 s21 Pa21 and H2/CO2 selectivity of 12.1 were obtained, which is promising in the potential application of H2 purification. The effects of H2O and CH3OH were examiend by experimental and simulation techniques. Both H2 permeance and H2/CO2 selectivity are almost unchanged by H2O, indicating that the pore in Zn (bdc)(ted)0.5 is not blocked by H2O. In contrast, notable decreases are detected in both H2 permeance and H2/CO2 selectivity in the presence of CH3OH. Specifically, H2 permeance decreases to 2 £ 1026 mol m22 s21 Pa21 and 100 H2/CO2 selectivity Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 Molecular Simulation ZIF-7 membrane20 ZIF-8 membrane21 ZIF-22 membrane23 ZIF-69 membrane33 ZIF-90 membrane22 MIL-53 membrane34 HKUST-1 membrane35 ZIF-95 membrane 10 MOFs Polymers 36 Upper bound 1991 37 Upper bound 2008 1 1 10 100 1000 10,000 1,00,000 H2 permeability / Barrers Figure 8. H2/CO2 selectivity versus H2 permeability in MOF and polymer membranes.[42] J. Jiang H2/CO2 selectivity decreases to 8.1. Due to the high hydrophobicity of Zn(bdc)(ted)0.5, CH3OH is strongly adsorbed and subsequently blocks the diffusion of rarely adsorbed H2, leading to the decrease in H2 permeance, and consequently the decrease in H2/CO2 selectivity. 7 cluster, CO2 diffusion is slowed down. Specifically, the diffusion coefficient of CO2 in PBI/ZIF-7 is approximately 30% of that in PBI. H2 and CO2 permeabilities in PBI/ZIF-7 membranes are greater than in neat PBI. Particularly, the higher the ZIF-7 loading, the greater is the permeability, and a slight enhancement in permselectivity. Polymer/MOF membranes. To improve the mechanical strength of polymer membranes, inorganic fillers such as zeolites and metal oxides are added to form mixed-matrix membranes (MMMs). However, inorganic fillers do not have good interfacial compatibility with polymer matrices, leading to a less ideal performance for gas separation. Recently, nano-sized ZIF-7 was dispersed into polybenzimidazole (PBI) to fabricate MMMs for H2 purification, and enhanced H2 permeability and H2/CO2 permselectivity were reported.[75] Both PBI and ZIF-7 contain the same organic unit namely benzimidazolate, such that PBI/ZIF-7 membranes could have a good interfacial compatibility. To facilitate a clear and deep microscopic understanding, we conducted atomistic simulation for H2/CO2 separation in PBI/ZIF-7 membranes.[44] Figure 9(a) demonstrates a membrane consisting of PBI and a two-cage ZIF-7 cluster. The void size distributions in pure PBI and PBI/ZIF-7 membranes are shown in Figure 9(b). With increased ZIF-7 ˚ ) increases, loading, the percentage of large voids (.3 A ˚ ) decreases. This while the percentage of small voids (0–3 A is attributed to the increased free volume (empty cage) upon adding ZIF-7 cluster. The modulus in PBI/ZIF-7 membrane gradually increases with increased ZIF-7 loading; thus, the mechanical strength is enhanced. This is a major advantage of MMMs over polymer membranes, which would allow MMMs to be practically used at high pressures. The solubility coefficients of both H2 and CO2 in PBI/ZIF-7 membranes increase with increased ZIF-7 loading. While H2 diffusion is not significantly affected by the presence of ZIF- Ionic liquid/MOF membranes. There has been a considerable interest in using supported ionic liquids (ILs) for carbon capture.[76,77] However, most studies to date have used either organic polymers or inorganic zeolites as supports with limited permeability. We have proposed IL/MOF membranes and demonstrated their use for CO2/N2 separation at room temperature. [45–47] In IRMOF-1-supported 1-n-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]), bulky [BMIM]þ cation resides in the open pore of IRMOF-1, whereas small [PF6]2 anion is located in metal cluster corner and possesses a strong interaction with the framework. [PF6]2 anion is the most favourable site for CO2 adsorption. With increasing IL ratio in the membrane, CO2/ N2 selectivity increases.[45] Moreover, ILs with four different anions including [PF6]2, tetrafluoroborate [BF4]2, bis(trifluoromethylsulphonyl)imide [Tf2N]2 and thiocyanate [SCN]2 were examined.[46] Among the four anions, [Tf2N]2 has the weakest interaction with IRMOF-1. CO2/N2 selectivity increases in the order of [Tf2N]2 , [PF6]2 , [BF4]2 , [SCN]2. This hierarchy largely follows the trend of binding energy between CO2 and anion estimated by ab initio method. [BMIM][SCN]/ IRMOF-1 outperforms polymer membranes and polymersupported ILs in CO2 permeability, and its performance surpasses Robeson’s upper bound as demonstrated in Figure 10. In addition, both hydrophobic ZIF-71 and hydrophilic Na-rho-ZMOF with identical topology and similar pore size were used as supports, and hydrophilic (a) (b) 20 pure PBI PBI/1-cage ZIF-7 PBI/2-cage ZIF-7 15 Percentage Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 524 PBI/3-cage ZIF-7 10 5 0 0 1 2 3 4 Void size (Å) 5 6 7 Figure 9. (Colour online) (a) A membrane of PBI/2-cage ZIF-7 cluster. (b) Void size distributions in pure PBI and PBI/ZIF-7 membranes.[44] Molecular Simulation 1000 [BMIM][SCN]/ZMOF Polymer-SILs Upper Bound PCO / PN 2 100 2 ILs/IRMOF-1 10 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 [BMIM][SCN]/ZIF-71 1 0.0001 0.01 1 100 4 10 PCO2 (barrer) Figure 10. (Colour online) CO2/N2 permselectivity versus CO2 permeability in IL/MOF membranes. The red circles are experimental data in polymer membranes and the line is Robeson’s upper bound. Also illustrated are the data in polymersupported ILs.[47] support was found to be superior to hydrophobic counterpart. [47] These simulation studies reveal that IL/MOF membranes are potentially intriguing for CO2 capture, and could trigger experimental efforts. 3.2 Hydrocarbon separation Hydrocarbons are predominantly used as combustible fuels and raw materials in chemical industry. Extensive simulation efforts have been conducted for hydrocarbons in zeolitic materials,[78] whereas the studies in MOFs are few. 3.2.1 Alkanes We simulated the adsorption and separation of linear and branched alkanes in IRMOF-1 at 300 K.[48] For pure linear alkanes (C1 to C5), a long alkane is more preferentially adsorbed than a short counterpart at low pressures, while the reverse is true at high pressures. At zero loading, the adsorption properties including isosteric heat, Henry’s constant and adsorption entropy exhibit linear relationships with alkane carbon number. For pure branched alkanes (C5 isomers), a linear isomer is more strongly adsorbed than its branched analogue. The adsorption capacities in IRMOF-1 are substantially greater than in silicalite and carbon nanotube bundle. For a fivecomponent mixture of C1 to C5 linear alkanes, the adsorption of long alkane first increases and then decreases with increasing pressure, while the adsorption of short alkane continually increases and progressively replaces 525 the long alkane at high pressures due to size entropy effect. For a three-component mixture of C5 isomers, the adsorption of each isomer increases with increasing pressure, and the branched isomer is less adsorbed due to configurational entropy effect. The adsorption and diffusion of alkane isomer mixtures (C4 and C5) were examined in catenated and non-catenated MOFs (IRMOF-13, IRMOF-14, PCN-6 and PCN-60 ) at 300 K.[49] As observed in IRMOF-1,[48] competitive adsorption also occurs here with a greater extent of adsorption for a linear isomer. An inflection is seen in the isotherm as a result of sequential adsorption at multiple favourable sites. Compared with non-catenated counterparts, IRMOF-13 and PCN-6 exhibit larger loading at low pressures because of constricted pores and stronger affinity; however, the reverse is true at high pressures due to smaller pore volume. Adsorption selectivity in the four MOFs is comparable with that in silicalite and carbon nanotube. As seen from the density contours in Figure 11, nC4 is preferentially adsorbed close to the metal oxide clusters in IRMOF-14, whereas inside the octahedral pockets in PCN-60 . The diffusivity of alkane decreases with the degree of branching because a slender isomer diffuses faster. In the presence of constricted pores, diffusivity in catenated MOFs is smaller than in noncatenated counterparts. However, the diffusion selectivity is larger in catenated MOFs. This study provides insightful microscopic mechanisms for the adsorption and diffusion of alkane isomers in MOFs, and reveals that both adsorption and diffusion selectivities can be enhanced by catenation. 3.2.2 Xylenes Xylenes are essential C8-aromatics derived from crude oil. The separation of xylene isomers ( p-, m- and o-xylenes) is practically important but challenging because of their similar physical properties. For example, the boiling points of p-, m- and o-xylenes are 138.4, 139.1 and 144.48C, and their kinetic diameters are 0.67, 0.71 and 0.74 nm, respectively.[79] We performed a non-equilibrium MD simulation to examine liquid chromatographic separation of xylene isomers in MIL-101.[50] MIL-101 acts as a stationary phase while hexane as a mobile phase, which was the case in the experiment.[80] Figure 12 plots the transport velocity of xylene versus external force aext acting on the mobile phase. The velocity of each isomer rises linearly with increasing a ext. The elution order follows p-xylene . m-xylene . o-xylene, which is independent of aext and in good agreement with experiment. For p-, mand o-xylenes, the interactions with MIL-101 (DEframework) increase from 2 24.6, 2 27.0 to 2 29.0 kJ/mol. Nevertheless, the interactions with hexane (DE solvent) J. Jiang Figure 11. (Colour online) Density contours of nC4 in (a) IRMOF-14 and (b) PCN-60 .[49] decrease slightly from 2 37.3, 2 36.9 to 2 36.5 kJ/mol. Consequently, p-xylene interacts the most weakly with MIL-101 and the most strongly with hexane, and has the fastest transport velocity. The opposite is in o-xylene, which has the slowest velocity. The DDE ( ¼ DEsolvent 2 DEframework) are 2 12.7, 2 9.9 and 2 7.5 kJ/mol, respectively, for p-, m- and o-xylenes. The DDE decreases in the order of p-xylene . m-xylene . o-xylene, which is consistent with the elution order. The difference in DDE among the three xylene isomers is greater than that in DEframework and DEsolvent. This implies that the separation of xylene isomers is attributed to the cooperative solute – framework and solute – solvent interactions. Analysis of radial distribution functions suggests that o-xylene is the closest to MIL-101, while p-xylene resides the farthest away. This is consistent with the energetic analysis that oxylene interacts the most strongly with MIL-101, as attributed to its largest polarity among the three xylene isomers. 4 3 v (m/s) Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 526 2 p-xylene m-xylene o-xylene 1 0.03 0.04 0.05 aext (nm/ps2) Figure 12. Transport velocity of xylene versus external force acting on mobile phase.[50] 3.3 Alcohol adsorption and biofuel purification Depletion of fossil fuels and increased CO2 concentration due to the combustion of fossil fuels have sparked a considerable interest worldwide to develop renewable energy resources such as biofuel. Compared with conventional fuels, biofuel is environmentally benign and carbon neutral with less emission of gaseous pollutants. As-produced biofuel contains a large amount of water and alcohol (e.g. ethanol), and it is a prerequisite to separate water/alcohol mixture to produce fuel-grade biofuel. The separation alone is estimated to account for 60 –80% of biofuel product cost [81]; thus, highly efficient biofuel purification is indispensable. A few experimental studies examined alcohol adsorption and biofuel purification in MOFs; nevertheless, simulation effort is rare. 3.3.1 Alcohol adsorption Combining experiment and simulation, we investigated methanol adsorption in Zn(bdc)(ted)0.5 and the simulated isotherm is in good agreement with the experiment [40]. In two MOFs (hydrophobic ZIF-71 and hydrophilic rhoZMOF) with identical topology and similar pore size, the adsorption of methanol and ethanol was examined.[51] The isotherms in Na-rho-ZMOF are type I, attributed to the high affinity of non-framework Naþ ions and ionic framework. In ZIF-71, the framework – adsorbate affinity is relatively weaker and type V adsorption is observed. The predicted ethanol adsorption in ZIF-71 matches fairly well with available experimental data. We further simulated the adsorption of a series of normal alcohols (C1 –C4) in ZIF-8.[53] Particularly, the effects of force fields and framework charges on alcohol adsorption were assessed. Although these effects have been, to a certain extent, examined for the adsorption of small gas molecules in ZIF-8, it is unknown or very Molecular Simulation 10 10 8 8 N (mmol/g) (b) 12 N (mmol/g) (a) 12 6 exp. UFF AMBER DREIDING 4 2 0 0 5 10 15 P (kPa) Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 6 4 exp. cluster charges periodic charges 2 20 527 0 0 5 10 15 20 P (kPa) Figure 13. (Colour online) (a) Effect of force fields on methanol adsorption. (b) Effect of framework charges on methanol adsorption. ‘cluster’ and ‘periodic’ indicate that the framework charges were estimated on the basis of cluster and periodic models, respectively.[53] limited on how they would affect alcohol adsorption. Figure 13 shows the adsorption isotherms of methanol in ZIF-8 at 308 K predicted by three force fields UFF, AMBER and DREIDING, as well as experimentally measured. With increasing pressure, the isotherm can be characterised into three regimes. At low pressures, adsorption extent is small corresponding to cluster formation at the favourable adsorption sites in ZIF-8. At intermediate pressures, cage filling occurs with a sharp increase in uptake. Finally, saturation is gradually approached at high-pressure regime. Among the three force fields, the uptake decreases in the order of UFF . AMBER . DREIDING. The predictions of DREIDING match the best with the experiment; particularly, the saturation loading is close to the measured value, although the loadings at low pressures are overestimated. The predictions using ‘cluster’ and ‘periodic’ charges are close except at intermediate pressures, and both match well with the experiment. This suggests that the interactions between methanol and ZIF-8 framework are dominated by the LJ potential, and the Coulombic potential plays a secondary role. The adsorption of ethanol, propanol and butanol in ZIF-8 was also simulated and compared with the experimental data. 3.3.2 Biofuel purification For biofuel purification, we simulated the separation of ethanol/water liquid mixtures in Na-rho-ZMOF and Zn4O (bdc)(bpz)2 at both pervaporation (PV, 508C) and vapour permeation (VP, 1008C) conditions.[52] In hydrophilic Narho-ZMOF, water is preferentially adsorbed over ethanol due to its strong interaction with non-framework Naþ ions and ionic framework, and the adsorption selectivity of water/ethanol is higher at a lower composition of water. With increasing water composition, the diffusion selectivity of water/ethanol increases. In contrast, ethanol is adsorbed more in hydrophobic Zn4O(bdc)(bpz)2 as attributed to the favourable interaction with methyl groups, and the adsorption selectivity of ethanol/water is higher at a lower composition of ethanol. With increasing ethanol composition, the diffusion selectivity of ethanol/ water increases slightly. The permselectivities in the two MOFs at both PV and VP conditions are largely determined by adsorption selectivity. In Na-rho-ZMOF, the maximum permselectivity is about 12 at VP condition, and Na-rho-ZMOF is preferable for biofuel dehydration. In Zn4O(bdc)(bpz)2, the maximum permselectivity is 75 at PV condition, and Zn4O(bdc)(bpz)2 is promising for biofuel recovery. This study provides microscopic insight into the separation of water/ethanol mixtures in hydrophilic and hydrophobic MOFs at both PV and VP conditions, and suggests that hydrophobic MOF is superior to hydrophilic counterpart for biofuel purification. Subsequently, simulation was conducted to screen hydrophobic MOFs including ZIF-8, 60, 71, 90, 96, towards high-performance purification of dilute biofuel at room temperature.[54] As shown in Figure 14, the simulated isotherm of ethanol for ethanol/water mixture in ZIF-8 matches fairly well with the experiment. Among the five MOFs, ZIF-8 is predicted to possess the highest ethanol/water selectivity. 3.4 Water treatment With increasing population, water scarcity has been a global concern and how to supply sufficient fresh water is a topical issue. On the other hand, a substantial amount of contaminants such as heavy metal ions and organic compounds have been introduced into water. These contaminants are toxic and carcinogenic, and thus, should be removed to minimise health and environmental risks. 528 J. Jiang (b) 50 (a) 7 5 4 3 2 exp. sim. 1 0 0.00 0.01 0.02 0.03 0.04 30 20 10 0.05 0 0.00 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 Xethanol Figure 14. 3.4.1 ZIF-8 ZIF-60 ZIF-71 ZIF-90 ZIF-96 40 Sethanol/water Nethanol (mmol/g) 6 0.01 0.02 0.03 Xethanol 0.04 0.05 (Colour online) (a) Adsorption of ethanol/water in ZIF-8 and (b) ethanol/water selectivity.[54] Water desalination More than 95% of water on the earth is seawater, which could supply abundant fresh water after economical desalination. A few desalination techniques such as reverse osmosis (RO) and thermal distillation have been developed for seawater.[82] In particular, RO accounts for half of the installed desalting capacity worldwide.[83] Nevertheless, the capital cost of current RO technology is high and needs to be improved prior to propagation. Membrane materials in RO play a pivotal role in desalting performance, and extensive studies have been reported towards the development of novel membranes ranging from polymeric membranes, carbon nanotubes to biological water channels (aquaporin).[84 –87] From non-equilibrium MD simulation, we demonstrated that ZIF-8 membrane could potentially act as an RO membrane. ZIF-8 was chosen because of its chemical/ thermal stability and remarkable resistance to water and organics. This is largely due to the hydrophobic cages in ZIF-8 and the strong tetrahedral ZnN4 clusters assembled by metals and linkers. The simulation system is schematically shown in Figure 15. Under external pressure, water desalination occurs through the ZIF-8 membrane, Figure 15. (Colour online) Water desalination through ZIF-8 membrane.[55] whereas Naþ and Cl2 ions cannot transport due to the sieving of small apertures in ZIF-8. The flux of water permeating the membrane scales linearly with external pressure. From temperature-dependent flux, the activation energy of water is estimated to be 24.4 kJ/mol. Because of surface interaction and geometrical confinement, water molecules in the ZIF-8 membrane experience less hydrogen bonding compared with bulk water. In addition, the life time of hydrogen bonding in the membrane is considerably longer. This simulation study suggests that ZIF-8 might be potentially useful for water desalination. 3.4.2 Removal of toxic ions As mentioned earlier, toxic ions in water should be removed for water treatment. In this context, we conducted simulation for the exchange of Pb2þ ions with Naþ ions in Na-rho-ZMOF at 298 K.[56] Pb2þ was chosen because it is toxic and a common contaminant after water transporting through lead-bearing household pipelines. Figure 16 shows the simulation system at different durations. Initially, Pb2þ and Cl2 ions reside in solution and Naþ ions are in rhoZMOF framework. Upon starting simulation, Pb2þ ions rapidly move into the framework while Naþ ions move out. At t ¼ 0.2 ns, a large number of Pb2þ ions have moved into rho-ZMOF, particularly near the solution/rho-ZMOF interface. Meanwhile, Naþ ions move out and stay in solution. A portion of Cl2 ions also move into the framework during ion exchange. Once exchanged, Pb2þ ions prefer staying in the framework without moving back to solution. This induces a Donnan effect on the distributions of Naþ and Cl2 ions between the framework and the solution. At t ¼ 2 ns, all Pb2þ ions are exchanged and reside in the framework. In addition, Pb2þ ions located at the solution/rho-ZMOF interface also move into rhoZMOF. By umbrella sampling, we estimated the potentials of mean force (PMFs) for ions moving from solution into Molecular Simulation 529 (a) t = 0 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 PbCl2 Na-rho-ZMOF (b) t = 0.2 ns PbCl2 (c) t = 2 ns Figure 16. (Colour online) Removal of Pd2þ by ion exchange in Na-rho-ZMOF: (a) t ¼ 0, (b) t ¼ 0.2 ns and (c) t ¼ 2 ns. Pb2þ: orange; Cl2: green; Naþ: blue.[56] rho-ZMOF. The PMF for Pb2þ is about 2 10 kBT and more favourable than 2 5 kBT for Naþ, which contributes to the observed ion exchange. The residence-time distributions and mean-squared displacements reveal that all the exchanged Pb2þ ions stay continuously in rho-ZMOF without exchanging with other ions in solution. Upon comparison, Naþ ions have a shorter residence time and a larger mobility. The exchanged Pb2þ ions in rho-ZMOF are located at the eight-, six- and four-membered rings. Due to different confinement effects, Pb2þ ions exhibit distinct dynamics at the three locations. Specifically, Pb2þ ions at the eight-membered ring have the highest mobility, while those at the four-membered ring have a negligible mobility. Quantitative understanding is provided by this study for ion exchange in ionic MOF and suggests that rho-ZMOF might be an intriguing candidate for the removal of toxic ions. 3.4.3 Removal of organics In chemical industries, organics are required to be removed/recovered from water. For example, dimethyl sulphoxide (DMSO) is often mixed with water to form biphasic medium, which is used to dissolve chemotherapeutic drug busulfan.[88]and formulate polymers, dyes and electronics.[89] To recycle, DMSO needs to be recovered from aqueous solution. Although distillation technology has been applied for DMSO recovery, it is energetically intensive. A simulation study for DMSO recovery from aqueous solution was reported in three hydrophobic MOFs, namely Zn4O(bdc)(bpz)2, Zn(bdc)(ted)0.5 and ZIF-71 at 298 K. [57] The free volumes in Zn4O(bdc)(bpz)2, Zn(bdc)(ted)0.5 and ZIF-71 are approximately 0.87, 0.79 and 0.44 cm3/g and the porosities are 69.4%, 64.9% and 50.4%, respectively. In the three MOFs, type I adsorption isotherms are observed for DMSO, while H2O exhibits type V adsorption isotherms with hysteresis. This implies a stronger interaction of DMSO than water with the three MOFs. The saturation capacity of DMSO is 10 mmol/g in Zn4O(bdc)(bpz)2, drops to 8.0 mmol/g in Zn(bdc)(ted)0.5 and 4.4 mmol/g in ZIF-71. Such a hierarchy is consistent with the decreasing order of free volume and porosity in the three MOFs. Due to the hydrophobic nature, the three MOFs are highly selective towards DMSO from DMSO/ H2O mixture. As shown in Figure 17, the selectivity in the three MOFs initially increases with increasing XDMSO, then decreases within the range of XDMSO under study. The initial increase is attributed to the enhanced interaction of DMSO with multiple adsorption sites in the framework. However, the interaction strength drops when most adsorption sites are occupied, and furthermore, smaller H2O molecules are more easily filled into the framework; consequently, a decrease in selectivity is seen at high 530 J. Jiang with MIL-101, and thus transports fastest. Furthermore, Arg forms the largest number of hydrogen bonds with water and possesses the largest hydrophilic solvent-accessible surface area. In contrast, Trp has the weakest interaction with water and is the closest to MIL-101. Elucidated from detailed energetic and structural analysis, the underlying separation mechanism is attributed to the cooperative solute – solvent and solute– framework interactions. The molecular insight from this simulation study suggests that MIL-101 might be an interesting material for the separation of important biomolecules. 1800 SDMSO/H2O 1500 1200 900 600 300 0 0.00 0.01 0.02 0.03 0.04 0.05 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 XDMSO Figure 17. (Colour online) DMSO/H2O selectivity in Zn4O (bdc)(bpz)2, Zn(bdc)(ted)0.5 and ZIF-71.[57] XDMSO. The highest selectivity is predicted up to 1700 in ZIF-71; nevertheless, the recovery capacity of DMSO in ZIF-71 is significantly smaller than in Zn4O(bdc)(bpz)2 and Zn(bdc)(ted)0.5. From a practical point of view, Zn4O (bdc)(bpz)2 and Zn(bdc)(ted)0.5 might be better among the three MOFs for DMSO recovery. 3.5 Bio- and chiral separation and drug loading 3.5.1 Bio-separation Bio-separation is of central importance in pharmaceutical industry, but experimental and simulation studies are scarcely reported for bio-separation using MOFs. In this regard, we conducted MD simulation to investigate the separation of amino acids (Arg, Phe and Trp) in MIL-101 at 300 K.[50] Similar to the liquid chromatographic separation of xylene isomers discussed earlier, MIL-101 is the stationary phase and water is the mobile phase, as illustrated in Figure 18. The three amino acids differ in structure, molecular weight and charge. The simulation reveals that the elution order is Arg . Phe . Trp. Among the three amino acids, Arg is the most hydrophilic. It experiences the strongest interaction with water but the weakest interaction Arg Phe MIL-101 Trp Figure 18. (Colour online) Separation of three amino acids (Arg, Phe and Trp) in MIL-101.[50] 3.5.2 Chiral separation In Mother Nature, living organisms exhibit significantly different biological responses to chiral enantiomers. One enantiomer may have desired pharmacological activity, while the other is inactive or toxic.[90] The production of pure enantiomeric compounds is crucial for health care. However, the separation of chiral molecules is challenging, as a pair of enantiomers display identical physical and chemical properties. Only in asymmetric environment, enantiomers experience different affinities and may be separated. Chiral separation by membrane is attractive because of its low cost, high capacity, continuous operation and easy scale-up. However, high-performance chiral separation membrane materials are currently scarce. By integrating experimental and simulation techniques, we examined the separation of racemic mixture of R/S-methyl phenyl sulphoxide (MPS) in an MOF membrane.[58] The MOF namely Zn2(bdc)(L-lac)(dmf) possesses a homochiral ˚ pore, and the chiral structure with approximately 5 A centres of L-lactate moieties are exposed along the pore. The membrane was prepared on ZnO support by reactive seeding method. The porous support acted as an inorganic source reacting with organic precursor to grow a seeding layer for the secondary growth of MOF membrane. Thus, a strong adhesive and uniform seeding layer was achieved to aid in the preparation of a superior MOF membrane. Experimentally, S-MPS was found to diffuse through the membrane in a relatively lower velocity than R-MPS. Parallel simulation predicted the same elution order, i.e. RMPS transports faster than S-MPS as shown in Figure 19 (a). To provide microscopic insight into the chiral separation, the interaction energies of R-/S-MPS with the MOF were estimated. As shown in Figure 19(b), S-MPS has a stronger interaction (in both LJ and Coulombic contributions) with the MOF than R-MPS. Therefore, R-MPS is less strongly bound onto the MOF and diffuses faster. This study potentially provides a new, sustainable and highly efficient technique for chiral separation. Molecular Simulation (b) –120 40 R-MPS S-MPS 20 –60 –30 10 0 0 0 Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 R-MPS S-MPS –90 30 ∆E (kJ/mol) Displacement (nm) (a) 531 2 4 6 8 LJ 10 Coulombic Total t (ns) Figure 19. (Colour online) (a) Displacements of R-/S-MPS in MOF as a function of simulation duration. (b) Interaction energies of R-/SMPS with MOF.[58] 3.5.3 Drug loading The interest in using nanoporous materials for drug loading/ delivery has increased considerably. The loading capacity and release of drug in a porous carrier are governed by a variety of factors such as pore size, shape, connectivity and host affinity. In conventional materials (e.g. inorganic silica, carbonaceous materials and polymeric matrixes), drug loading capacity is usually not sufficiently high and encapsulated drug is difficult to be specifically released. To achieve a high loading and a controlled release, porous materials such as MOFs possessing large volumes and regular structures are desired. We report a simulation study to understand the microscopic interactions of ibuprofen (IBU) with MIL-101 and UMCM-1.[59] IBU was chosen as a model drug because it is commonly used for the relief of arthritis, dysmenorrhea, acne and fever. MIL-101 and UMCM-1 possess large pore volumes (1.96 and 2.28 cm3/g) and high surface areas (3451 and 4764 m2/g). Particularly, MIL-101 contains unsaturated Lewis acidic metal sites by removing terminal waters in Figure 20. octahedral Cr3O trimmers; however, UMCM-1 does not have. The predicted maximum loading of IBU in MIL-101 at 298 K is 1.11 g IBU/g MIL-101, which agrees fairly well with experimentally determined 1.37. In both MIL-101 and UMCM-1, IBU loading is four times higher than in silica MCM-41.[91] The substantially high loading in the two MOFs implies that they might be useful for drug delivery as a small amount of carrier is needed for high dosage. To assess possible bond formation between IBU and host structure, the highest occupied molecular orbitals (HOMOs) were calculated.[59] As shown in Figure 20, the HOMOs are distinctly different between MIL-101 and UMCM-1. From the frontier HOMOs, a coordination bond appears to form between the carboxylic group in IBU and the Cr3O metal oxide in MIL-101. The band gap is only 0.064 eV in IBU/MIL-101, significantly lower than 3.146 eV in IBU/UMCM-1. The latter is prohibitively large and implies difficulty for IBU and UMCM-1 to form a bond. Based on the mean-squared displacement, the mobility of IBU in MIL-101 is substantially smaller than (Colour online) HOMOs in IBU/MIL-101 and IBU/UMCM-1.[59] J. Jiang Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 in UMCM-1. This is attributed to the coordination bond formation between IBU and MIL-101, which is the primary factor for the delayed release of IBU in MIL-101 experimentally observed. This study unravels the energetics and dynamics of IBU in two mesoscopic MOFs at a molecular level. Nevertheless, we should note that the experimental study of IBU loading/delivery in MIL-101 was conducted in a solvent (hexane). However, solvent was not included in our simulation. The inclusion of solvent would screen the interaction and reduce the accessibility between drug and carrier. Thus, more elaborate investigation on drug behaviour in MOFs should take solvent into account. 0.4 0.2 Stress sv (GPa) 532 0.0 –0.2 simulation experiment –0.4 –0.06 –0.03 0.00 0.03 0.06 strain ev Figure 21. ZIF-8.[60] (Colour online) Volumetric stress versus strain in 3.6 Mechanical moduli, structural transition and thermal conductivity Most simulation studies in MOFs assume that the frameworks are fixed. However, many MOFs exhibit framework flexibility. For example, gas molecules (N2, CH4, C2H6 and C3H8) with kinetic diameters larger than the window size of ZIF-8 were observed to diffuse through ZIF-8.[92 – 94] Therefore, the framework flexibility should be incorporated to properly describe such MOFs. For the prototype ZIF-8, we have developed force fields by combining DFT calculation, Amber force field and experimental crystallographic data.[60,61] The force fields were adopted to predict the mechanical moduli, structural transition and thermal conductivity of ZIF-8. 5.35 and 5.94 GPa upon introducing one-cage, two-cage and three-cage ZIF-7 clusters, respectively.[44] Therefore, the modulus in PBI/ZIF-7 membrane is gradually enhanced with increased ZIF-7 loading. It is noteworthy that mechanical properties are sensitive to force field used and their accurate prediction is a challenging task. Based on classical force fields (e.g. DREIDING, CVFF and MM3), a few simulation studies overestimated Young’s and bulk moduli of MOF-5 by several folds.[98 – 100] Different force fields even gave drastically different moduli, indicating their incapability to quantitatively describe the mechanical properties. 3.6.1 Mechanical moduli For solid materials, mechanical strength can be quantified by moduli.[95] By including framework flexibility, a force field developed for ZIF-8 was used to estimate the bulk and Young’s moduli.[60] Figure 21 plots the volumetric stress sv versus strain 1v in ZIF-8. The relationship of simulated sv and 1v is approximately linear from 2 0.3 to 0.3 GPa, though the experimental stress –strain curve is nonlinear at high pressures. The bulk modulus Ev predicted is about 7.24 ^ 0.03 GPa, close to experimentally determined 6.52 ^ 0.35 GPa at low pressures.[96] Similarly, the stress along c direction sc also increases linearly with the strain 1c . On this basis, Young’s modulus Ec is estimated to be 2.99 ^ 0.08 GPa, which agrees well with an experimental value of 2.97 ^ 0.06 GPa.[97] Young’s modulus in the a or b direction is nearly identical to Ec, implying that ZIF-8 is mechanically isotropic. This prediction is in accord with the fact that ZIF-8 is a cubic crystal and has homogeneous atomic distribution along three orthogonal directions. As discussed earlier, one major advantage of MMMs over polymer membranes is improved mechanical strength. In PBI/ZIF-7 membranes, our simulation reveals that the bulk modulus of PBI increases from 4.11 to 4.77, 3.6.2 Structural transition Upon external stimuli (e.g. temperature, pressure and sorption), some MOFs may undergo structural transition. Understanding the fundamental mechanism of structural transition is crucial for their potential applications in sensing, separation, drug delivery, etc. However, it is experimentally difficult to examine structural transition microscopically. By developing a hybrid MC/MD simulation method, we successfully mimic the continuous structural transition of ZIF-8 induced by N2 sorption at 77 K.[61] Unlike the commonly used GCMC method in which framework is fixed, ZIF-8 in the hybrid MC/MD method is allowed to relax along with N2 sorption. As shown in Figure 22, stepped sorption isotherm is predicted with three distinct regions, which agrees well with experimental data. Three regions correspond to a high-loading (HL) structure, a lowloading (LL) structure and the transition between HL and LL. Upon structural transition, the cell size rises, the orientation of imidazolate rings and the motion of framework atoms exhibit sharp changes. In addition, pronounced changes occur in various contributions of potential energies (including stretching, bending, torsional, van der Waals and Coulombic). Radial distribution functions reveal strong Molecular Simulation N2 (molecules/uc) 50 experiment simulation 40 30 20 10 0 10–5 10–4 10–3 10–2 10–1 100 Figure 22. (Colour online) Structural transition of ZIF-8 induced by N2 sorption.[61] interactions between N2 and imidazolate rings. The simulation suggests that the structural transition of ZIF-8 is caused by the reorientation of imidazolate rings, which is attributed to the enhanced van der Waals interaction between sorbed N2 and imidazolate rings as well as the reduced torsional interaction in framework. This study provides clear and quantitative microscopic understanding for the structural transition of ZIF-8, and has an important implication for other porous materials. 3.6.3 Thermal conductivity Adsorption/desorption in MOFs is exothermic/endothermic process. The heat released/adsorbed would affect system temperature and process performance. For instance, heat transfer is associated with H2 storage system and appropriate thermal management is required to retain energy efficiency.[101] Furthermore, heat transfer in nanoporous materials is also substantially important for (a) 0.20 the development of electronic, optical and optoelectronic devices.[102] To our best knowledge, only one simulation study was reported in the literature to examine the thermal conductivity of MOF-5.[103] Based on a force field we developed,[60] thermal conductivity of ZIF-8 and corresponding mechanism were investigated.[62] The simulated conductivity is about 0.165 W/mK. Such a low thermal conductivity is due to the short mean free path of phonon in ZIF-8, which is estimated to be less than two unit cells. As shown in Figure 23, the conductivity increases from 0.165 to 0.190 W/mK with an increasing temperature from 300 to 1000 K. The temperature effect is attributed to the enhanced overlap in the vibrational density of states between Zn and N atoms. The contributions of lattice vibrations in different directions to heat flux were examined from non-equilibrium MD simulation. It was found that the longitudinal vibration contributes 60% to thermal transport in ZIF-8; in contrast, transverse vibration contributes 40%. Furthermore, the contributions from different forces to heat flux were analysed. While the stretching and Lennard-Jones components have 31% contribution each, the bending and Coulombic components contribute 17% and 21%, respectively. However, the contribution of torsional component is nearly zero. Although this study is focused on ZIF-8, the methodology can be extended to other ZIFs and MOFs. 4. Summary and perspective This manuscript summarises the comprehensive and systematic simulation endeavours in the author’s research group for diverse potential applications of MOFs, including carbon capture, hydrocarbon separation, alcohol adsorption and biofuel purification, water treatment, bioand chiral separation, drug loading, as well as mechanical moduli, structural transition and thermal conductivity. (b) 0.8 Zn N overlap 0.19 0.6 0.18 VDOS Thermal conductivity (W/mK) Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 P/P0 533 0.17 0.4 0.2 0.16 0.15 0.0 300 400 500 600 700 800 Temperature (K) 900 1000 0 10 20 30 40 50 60 Frequency (THz) Figure 23. (Colour online) (a) Temperature effect on the thermal conductivity of ZIF-8. (b) Vibrational density of states for Zn and N atoms and their overlap at 300 K.[62] Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 534 J. Jiang While most simulations in MOFs have been focused on gas storage and separation, our group has initiated several simulation studies for potential applications in liquid separation (e.g. biofuel purification, water treatment and bio- and chiral separation). With increasing demands for clean water, liquid fuels and high-purity biomolecules, it is foreseeable that more simulation studies will be conducted for liquid separation in the near future. A wealth of atomic-resolution and time-resolved insights have been provided by simulation to critically understand microscopic properties (both equilibrium and dynamic) in MOFs. These molecular insights are not just important complement to experiments, but essential to secure correct interpretation of experimental data and to test hypothetical candidates. Subsequently, MOFs can be screened from numerous candidates in a high-throughput manner or rationally designed by incorporating specific functionality, with little resort to costly and lengthy experimental testing. Despite the considerable progress in quantitative mechanistic understanding of MOFs, we should note that a number of challenges exist for future simulation exploration. (1) Most simulation studies in MOFs adopt empirical or semi-empirical force fields. While certain success is achieved, these force fields may lead to inaccurate predictions as they were constructed with empirical rules. In this regard, a general and transferable force field is highly desirable. Nevertheless, developing such a force field for MOFs is technically challenging because a wide variety of metal clusters and organic linkers are involved in MOFs. (2) Mechanical strength of MOFs is crucial for practical applications. In gas storage and separation, as well as liquid separation, the pressure exerted on MOF sorbents or membranes may distort structure and deteriorate performance. It is important to understand how pressure affects framework and pore geometry. Currently, only limited simulation studies estimate mechanical moduli. As discussed earlier, the mechanical properties of MOFs are sensitive to force field used and difficult to be accurately predicted. (3) A large number of MOFs are chemically unstable in water and organic solvents, which impedes their practical use in moisture or liquid phase. Furthermore, thermal stability should also be taken into account in applications, e.g. high-temperature pre-combustion carbon capture. Fundamental understanding in the chemical and thermal stability of MOFs is indispensable. It is useful to unravel by simulation the important factors that govern stability, thus providing guidelines on the selection of suitable building blocks to synthesise stable MOFs. However, simulation studies on this topic are rare and more efforts are needed. To recapitulate, molecular simulation has played an increasingly important role in the vibrant field of MOFs. Significant progress has been achieved; however, challenges still exist and indeed provide new opportunities for future simulation studies to unravel more in-depth microscopic insights and thus provide bottom-up guidelines on the rational screening and design of novel MOFs. For practical applications, both process requirements and material properties should be considered. Therefore, it is imperative to holistically synergise simulation with process optimisation, as well as material synthesis, towards the development of best MOFs for advanced technological innovation. Acknowledgements The author gratefully acknowledges his coworkers and collaborators for their contributions to the studies reviewed here and the National University of Singapore, the Ministry of Education of Singapore and the Singapore National Research Foundation for financial support. References [1] Yaghi OM, O’Keefe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J. Reticular synthesis and design of new materials. Nature. 2003;423:705 –714. [2] Fe´rey G. Hybrid porous solids: past, present, future. Chem Soc Rev. 2008;37:191–214. [3] Czaja AU, Trukhan N, Muller U. Industrial applications of metal – organic frameworks. Chem Soc Rev. 2009;38: 1284–1293. [4] Meek ST, Greathouse JA, Allendorf MD. Metal – organic frameworks: a rapidly growing class of versatile nanoporous materials. Adv Mater. 2011;23:249–267. [5] Adams J, Pendlebury D. Global research report: Materials science and technology. Leeds: Thomson Reuters; 2011. [6] Suh MP, Park HJ, Prasad TK, Lim DW. Hydrogen storage in metal–organic frameworks. Chem Rev. 2012;112:782–835. [7] Sumida K, Rogow DL, Mason JA, McDonald TM, Block ED, Herm ZR, Bae TH, Long JR. Carbon dioxide capture in metal– organic frameworks. Chem Rev. 2012;112:724 –781. [8] Hill JR, Subramanian L, Maiti A. Molecular modeling techniques in material sciences. London: Taylor & Francis; 2005. [9] Duren T, Bae YS, Snurr RQ. Using molecular simulation to characterise metal–organic frameworks for adsorption applications. Chem Soc Rev. 2009;38:1203– 1212. [10] Liu DH, Zhong CL. Understanding gas separation in metal– organic frameworks. J Mater Chem. 2010;20:10308–10318. [11] Jiang JW, Babarao R, Hu ZQ. Molecular simulations for energy, environmental and pharmaceutical applications of nanoporous materials: from zeolites, metal–organic frameworks to protein crystals. Chem Soc Rev. 2011;40:3599–3612. [12] Jiang JW. Metal–organic frameworks for CO2 capture: what are learned from molecular simulations. In: Ortiz OL, Ramı´rez LD, editors. Coordination polymers and metal organic frameworks. New York: Nova Science Publishers; 2012. [13] Jiang JW. Recent development of in silico molecular modeling for gas and liquid separations in metal–organic frameworks. Curr Opin Chem Eng. 2012;1:138 –144. [14] Getman RB, Bae YS, Wilmer CE, Snurr RQ. Review and analysis of molecular simulations of methane, hydrogen and acetylene storage in metal–organic frameworks. Chem Rev. 2012;112: 703–723. [15] Campan˜´ C, Mussard B, Woo TK. Electrostatic potential derived atomic charges for periodic systems using a modified error functional. J Comput Chem. 2009;5:2866–2878. [16] Mans TA, Sholl DS. Chemically meaningful atomic charges that reproduce the electrostatic potential in periodic and nonperiodic materials. J Chem Theory Comput. 2010;6:2455–2468. [17] Xu Q, Zhong CL. A general approach for estimating framework charges in metal – organic frameworks. J Phys Chem C. 2010;114:5035–5042. Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 Molecular Simulation [18] Wilmer CE, Kim KC, Snurr RQ. An extended charge equilibration method. J Phys Chem Lett. 2013;3(2):2506– 2511. [19] Jiang JW, Sandler SI. Separation of CO2 and N2 by adsorption in C168 schwarzite: a combination of quantum mechanics and molecular simulation study. J Am Chem Soc. 2005;127: 11989– 11997. [20] Jiang JW, Klauda JB, Sandler SI. Hierarchical modeling N2 adsorption on the surface of and within a C60 crystal: from quantum mechanics to molecular simulation. J Phys Chem B. 2005;109:4731–4737. [21] Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. Uff: a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc. 1992;114:10024–10035. [22] Mayo SL, Olafson BD, Goddard WA. Dreiding: a generic force field for molecular simulations. J Phys Chem. 1990;94: 8897– 8909. [23] Allen MP, Tildesley DJ. Computer simulation of liquids. Oxford: Oxford University Press; 1987. [24] Frenkel D, Smit B. Understanding molecular simulation: from algorithms to applications. San Diego, CA: Academic Press; 2002. [25] Babarao R, Hu ZQ, Jiang JW, Chempath S, Sandler SI. Storage and separation of CO2 and CH4 in silicalite, C168 schwarzite, and IRMOF-1: a comparative study from monte carlo simulation. Langmuir. 2007;23:659–666. [26] Babarao R, Jiang JW, Sandler SI. Molecular simulations for adsorptive separation of CO2/CH4 mixture in metal-exposed, catenated, and charged metal–organic frameworks. Langmuir. 2009;25:5239 –5247. [27] Chen YF, Jiang JW. A bio-metal–organic framework for highly selective CO2 capture: a molecular simulation study. ChemSusChem. 2010;3:982–988. [28] Panda T, Pachfule P, Chen YF, Jiang JW, Banerjee R. Amino functionalized zeolitic tetrazolate framework with high capacity for storage of CO2. Chem Commun. 2011;47:2011 –2013. [29] Pachfule P, Chen YF, Jiang JW, Banerjee R. Experimental and computational approach of understanding the gas adsorption in amino functionalized interpenetrated metal organic frameworks. J Mater Chem. 2011;21:17737–17745. [30] Pachfule P, Chen YF, Sahoo SC, Jiang JW, Banerjee R. Structural isomerism and effect of fluorination on gas adsorption in coppertetrazolate based metal organic frameworks. Chem Mater. 2011;23:2908 –2916. [31] Pachfule P, Chen YF, Jiang JW, Banerjee R. Fluorinated metal– organic frameworks: advantageous for higher H2 and CO2 adsorption or not?. Chem-Eur J. 2012;18:688–694. [32] Zhang K, Chen YF, Nalaparaju A, Jiang JW. Functionalized metal–organic framework MIL-101 for CO2 capture: multi-scale modeling from ab initio calculation, molecular simulation to breakthrough prediction, submitted for publication 2013. [33] Jiang JW. Charged soc metal–organic framework for highefficacy H2 adsorption and syngas purification: atomistic simulation study. AIChE J. 2009;55:2422 –2432. [34] Babarao R, Jiang JW. Unprecedentedly high selective adsorption of gas mixtures in rho zeolite-like metal– organic framework: a molecular simulation study. J Am Chem Soc. 2009;131: 11417– 11425. [35] Babarao R, Eddaoudi M, Jiang JW. Highly porous ionic rht metal – organic framework for H2 and CO2 storage and separation: a molecular simulation study. Langmuir. 2010;26: 11196– 11203. [36] Babarao R, Jiang JW. Cation characterization and CO2 capture in Liþ-exchanged metal–organic frameworks: from first-principles modeling to molecular simulation. Ind Eng Chem Res. 2011;50:62 –68. [37] Chen YF, Nalaparaju A, Eddaoudi M, Jiang JW. CO2 adsorption in mono-, di- and trivalent cation-exchanged metal–organic frameworks: a molecular simulation study. Langmuir. 2012;28:3903 –3910. [38] Babarao R, Jiang JW. Upgrade of natural gas in rho zeolite-like metal–organic framework and effect of water: a computational study. Energy Environ Sci. 2009;2:1088– 1093. 535 [39] Chen YF, Babarao R, Sandler SI, Jiang JW. Metal organic framework MIL-101 for adsorption and effect of terminal water molecules: from quantum mechanics to molecular simulation. Langmuir. 2010;26:8743 –8750. [40] Chen YF, Lee JY, Babarao R, Li J, Jiang JW. A highly hydrophobic metal organic framework Zn(bdc)(ted)0.5 for adsorption and separation of CH3OH/H2O and CO2/CH4: an integrated experimental and simulation study. J Phys Chem C. 2010;114:6602–6609. [41] Babarao R, Jiang JW. Diffusion and separation of CO2 and CH4 in silicalite, C168 schwarzite, and IRMOF-1: a comparative study from molecular dynamics simulation. Langmuir. 2008;24: 5474–5484. [42] Huang AS, Chen YF, Wang NY, Hu ZQ, Jiang JW, Caro J. A highly permeable and selective zeolitic imidazolate framework ZIF-95 membrane for H2/CO2 separation. Chem Commun. 2012;48:10981–10983. [43] Huang AS, Chen YF, Liu Q, Wang NY, Jiang JW, Caro J. Synthesis of highly hydrophobic and permselective metal– organic framework Zn(bdc)(ted)0.5 membranes for H2/CO2 separation, submitted for publication 2013. [44] Zhang LL, Hu ZQ, Jiang JW. Metal– organic framework/polymer mixed-matrix membranes for H2/CO2 separation: a fully atomistic simulation study. J Phys Chem C. 2012;116: 19268–19277. [45] Chen YF, Hu ZQ, Gupta KM, Jiang JW. Ionic liquid/metal – organic framework composite for CO2 capture: a computational investigation. J Phys Chem C. 2011;115:21736–21742. [46] Gupta KM, Chen YF, Hu ZQ, Jiang JW. Metal –organic framework supported ionic liquid membranes for CO2 capture: anion effects. Phys Chem Chem Phys. 2012;14:5785–5794. [47] Gupta KM, Chen YF, Jiang JW. Ionic liquid membranes supported by hydrophobic and hydrophilic metal – organic frameworks for CO2 capture. J Phys Chem C. 2013;117: 5792–5799. [48] Jiang JW, Sandler SI. Monte carlo simulation for the adsorption and separation of linear and branched alkanes in IRMOF-1. Langmuir. 2006;22:5702 –5707. [49] Babarao R, Tong YH, Jiang JW. Molecular insight into adsorption and diffusion of alkane isomer mixtures in metal –organic frameworks. J Phys Chem B. 2009;113:9129–9136. [50] Hu ZQ, Chen YF, Jiang JW. Liquid chromatographic separation in metal–organic framework MIL-101: a molecular simulation study. Langmuir. 2013;29:1650 –1656. [51] Nalaparaju A, Zhao XS, Jiang JW. Molecular understanding for the adsorption of water and alcohols in hydrophilic and hydrophobic zeolitic metal– organic frameworks. J Phys Chem C. 2010;114:11542– 11550. [52] Nalaparaju A, Zhao XS, Jiang JW. Biofuel purification by pervaporation and vapor permeation in metal–organic frameworks: a computational study. Energy Environ Sci. 2011;4: 2107–2116. [53] Zhang K, Zhang LL, Jiang JW. Adsorption of C1 –C4 alcohols in zeolitic imidazoalte framework-8, submitted for publication 2013. [54] Nalaparaju A, Chen YF, Jiang JW. Computional screening of metal–organic frameworks for biofuel purification, submitted for publication 2013. [55] Hu ZQ, Chen YF, Jiang JW. Zeolitic imidazolate framework-8 as a reverse osmosis membrane for water desalination: insight from molecular simulation. J Chem Phys. 2011;134:134705. [56] Nalaparaju A, Jiang JW. Ion exchange in metal – organic framework for water purification: insight from molecular simulation. J Phys Chem C. 2012;116:6925–6931. [57] Nalaparaju A, Jiang JW. Recovery of dimethyl sulfoxide from aqueous solutions by highly selective adsorption in hydrophobic metal–organic frameworks. Langmuir. 2012;28:15305–15312. [58] Wang WJ, Dong XL, Nan JP, Jin WQ, Hu ZQ, Chen YF, Jiang JW. A homochiral metal–organic framework membrane for enantioselective separation. Chem Commun. 2012;48: 7022–7024. Downloaded by [NUS National University of Singapore] at 16:05 05 February 2014 536 J. Jiang [59] Babarao R, Jiang JW. Unraveling the energetics and dynamics of ibuprofen in mesoporous metal–organic frameworks. J Phys Chem C. 2009;113:18287–18291. [60] Hu ZQ, Zhang LL, Jiang JW. Development of a force field for zeolitic imidazolate framework-8 with structural flexibility. J Chem Phys. 2012;136:244703. [61] Zhang LL, Hu ZQ, Jiang JW. Sorption-induced structural transition of zeolitic imidazolate framework-8: a hybrid molecular simulation study. J Am Chem Soc. 2013;135: 3722–3728. [62] Zhang XL, Jiang JW. Thermal conductivity of zeolitic imidazolate framework-8: a molecular simulation study. J Phys Chem C, submitted for publication 2013. [63] Maginn EJ. What to do with CO2? J Phys Chem Lett. 2010;1:3478–3479. [64] Figueroa JD, Fout T, Plasynski S, Mcllvried H, Srivastava RD. Advances in CO2 capture technology—US Department of energy’s carbon sequestration program. Int J Greenhouse Gas Control. 2008;2:9–20. [65] Keskin S, van Heest TM, Sholl DS. Can metal – organic framework materials play a useful role in large-scale carbon dioxide separations? ChemSusChem. 2010;3:879–891. [66] Fe´rey G, Serre C, Devic T, Maurin G, Jobic H, Llewellyn PL, Weireld GD, Vimont A, Daturi M, Chang JS. Why hybrid porous solids capture greenhouse gases?. Chem Soc Rev. 2011;40: 550–562. [67] Li JR, Ma YG, McCarthy MC, Sculley J, Yu JM, Jeong HK, Balbuena PB, Zhou HC. Carbon dioxide capture-related gas adsorption and separation in metal–organic frameworks. Coord Chem Rev. 2011;255:1791– 1823. [68] Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O’Keefe M, Yaghi OM. Systematic design of pore size and functionality in isoreticular metal–organic frameworks and their application in methane storage. Science. 2002;295:469–472. [69] Myers AL, Prausnitz JM. Thermodynamics of mixed-gas adsorption. AIChE J. 1965;11:121–127. [70] Ma SQ, Sun DF, Ambrogio M, Fillinger JA, Parkin S, Zhou HC. Framework-catenation isomerism in metal–organic frameworks and its impact on H2 uptake. J Am Chem Soc. 2007;129: 1858– 1859. [71] Fe´rey G, Mellot-Draznieks C, Serre C, Millange F, Dutour J, Surble´ S, Margiolaki I. A chromium terephthalate-based solid with unusually large pore volumes and surface area. Science. 2005;309:2040–2042. [72] Liu YL, Kravtsov V, Larsen R, Eddaoudi M. Molecular building blocks approach to the assembly of zeolite-like metal–organic frameworks with extra-large cavities. Chem Commun. 2006;42:1488 –1490. [73] Betard A, Fischer RA. Metal–organic framework thin films: From fundamentals to applications. Chem Rev. 2012;112: 1055– 1083. [74] Robeson LM. The upper bound revisited. J Membr Sci. 2008;320:390 –400. [75] Yang TX, Xiao YC, Chung TS. Poly-/metal-benzimidazole nanocomposite membranes for hydrogen purification. Energy Environ Sci. 2011;4:4171– 4180. [76] Noble RD, Gin DL. Perspective on ionic liquids and ionic liquid membranes. J Membr Sci. 2011;369:1 –4. [77] Lozano LJ, Godinez C, de los Rios AP, Hernandez-Fernandez FJ, Sanchez-Segado S, Alguacil FJ. Recent advances in supported ionic liquid membrane technology. J Membr Sci. 2011;376:1 –14. [78] Smit B, Maesen TLM. Molecular simulations of zeolites: adsorption, diffusion, and shape selectivity. Chem Rev. 2008;108:4125–4184. [79] Fabri J, Graeser U, Simo T. Xylenes: Ullmann’s encyclopedia of industrial chemistry. 6th ed. Weinheim: Wiley-VCH; 2002. [80] Yang CX, Yan XP. Metal–organic framework MIL-101 for highperformance liquid chromatographic separation of substituted aromatics. Anal Chem. 2011;83:7144–7150. [81] Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T. The path [82] [83] [84] [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] [95] [96] [97] [98] [99] [100] [101] [102] [103] forward for biofuels and biomaterials. Science. 2006;311: 484–488. Khawaji AD, Kutubkhanah IK, Wie JM. Advances in seawater desalination technologies. Desalination. 2008;221:47–69. Shannon MA, Bohn PW, Elimelech M, Georgiadis JG, Marinas BJ, Mayes AM. Science and technology for water purification in the coming decades. Nature. 2008;452:301 –310. Li D, Wang HT. Recent developments in reverse osmosis desalination membranes. J Mater Chem. 2010;20:4551–4566. Hilder TA, Gordon D, Chung SH. Salt rejection and water transport through boron nitride nanotubes. Small. 2009;5: 2183–2190. Kumar M, Grzelakowski M, Zilles J, Clark M, Meier W. Highly permeable polymeric membranes based on the incorporation of the functional water channel protein aquaporin-Z. Proc Natl Acad Sci USA. 2007;104:20719–20724. Corry B. Designing carbon nanotube membranes for efficient water desalination. J Phys Chem B. 2008;112:1427–1434. Bhagwatwar HP, Phadungpojna S, Chow DS, Andersson BS. Formulation and stability of busulfan for intravenous administration in high-dose chemotherapy. Cancer Chemother Pharmacol. 1996;37:401–408. Park SJ, Yoon TI, Bae JH, Seo HJ, Park HJ. Biological treatment of wastewater containing dimethyl sulphoxide from the semiconductor industry. Process Biochem. 2001;36:579– 589. Lough WL, Wainer IW. Chirality in natural and applied science. London: Wiley-Blackwell; 1991. Vallet-Regi M, Balas F, Arcos D. Mesoporous materials for drug delivery. Angew Chem Int Ed. 2007;46:7548 –7558. Bux H, Liang FY, Li YS, Cravillon J, Wiebcke M, Caro J. Zeolitic imidazolate framework membrane with molecular sieving properties by microwave-assisted solvothermal synthesis. J Am Chem Soc. 2009;131:16000–16001. Gucuyener C, van den Bergh J, Gascon J, Kapteijn F. Ethane/ ethene separation turned on its head: selective ethane adsorption on the metal–organic framework ZIF-7 through a gate-opening mechanism. J Am Chem Soc. 2010;132:17704–17706. Zhang C, Lively RP, Zhang KZ, Johnson JR, Karvan O, Koros WJ. Unexpected molecular sieving properties of zeolitic imidazolate framework-8. J Phys Chem Lett. 2012;3: 2130–2134. Tan JC, Cheetham AK. Mechanical properties of hybrid inorganic-organic framework materials: establishing fundamental structure – property relationships. Chem Soc Rev. 2011;40: 1059–1080. Chapman KW, Halder GJ, Chupas PJ. Pressure-induced amorphization and porosity modification in a metal–organic framework. J Am Chem Soc. 2009;131:17546–17547. Tan JC, Bennett TD, Cheetham AK. Chemical structure, network topology, and porosity effects on the mechanical properties of zeolitic imidazolate frameworks, Proc. Natl Acad Sci USA. 2010;107:9938–9943. Kuc A, Enyashin A, Seifert G. Metal–organic frameworks: structural, energetic, electronic, and mechanical properties. J Phys Chem B. 2007;111:8179–8186. Greathouse JA, Allendorf MD. Force field validation for molecular dynamics simulations of IRMOF-1 and other isoreticular zinc carboxylate coordination polymers. J Phys Chem C. 2008;112:5795–5802. Tafipolsky M, Schmid R. Systematic first principles parameterization of force fields for metal–organic frameworks using a genetic algorithm approach. J Phys Chem B. 2009;113: 1341–1352. Zhang J, Fisher TS, Ramachandran PV, Gore JP, Mudawar I. A review of heat transfer issues in hydrogen storage technologies. J Heat Transfer. 2005;127:1391–1399. Luo TF, Chen G. Nanoscale heat transfer – from computation to experiment. Phys Chem Chem Phys. 2013;15: 3389–3412. Huang BL, McGaughey AJH, Kaviany M. Thermal conductivity of metal– organic framework-5: molecular dynamics simulations. Int J Heat Mass Transfer. 2007;50:393– 404.
© Copyright 2026 ExpyDoc
![Comment on “New Calix[4]arene Appended Amberlite XAD‑4 Resin](http://s3.expydoc.com/store/data/001467637_1-39f813bbb18f112c4010a3c970bd0c81-250x500.png)